


Our research program is focused on uncovering how the human transcriptome is regulated in the context of health and disease. Dynamic regulation of the transcriptome is central to the development and maintenance of an organism's phenotype. This includes morphological features, physiological attributes, and susceptibility to diseases. We aim to understand how changes in the transcriptome explain phenotypic differences between individuals including those with age, sex, genetic ancestry, and disease susceptibility. Understanding the molecular drivers of human phenotypic variation is crucial for treating patients according to their individual needs rather than generalized disease categories. Thus, our research has the ultimate goal of advancing the biomedical field towards personalized medicine.
Objectives
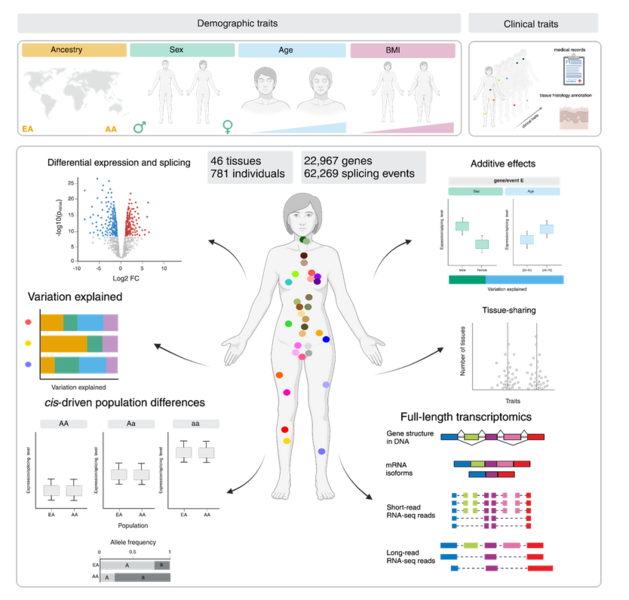
Research Line 2: Transcriptional regulation at single-cell resolution
Inter-individual differences in gene expression observed at the tissue level can arise from tissue-specific expression changes, or cell type composition shifts. Single-cell technologies are a powerful tool to disentangle the contribution of each component by profiling the transcriptome of individual cells. The expanded scale of single-cell techniques now also offer the opportunity to investigate how donor characteristics (such as sex, age, genetic ancestry, or body mass index) or environmental conditions (such as immune stimulation or lifestyle) shape cellular composition, gene expression, chromatin accessibility, and alternative splicing at an unprecedented resolution.
Since January 2020, we are part of the steering committee of the single-cell eQTL Consortium, (https://eqtlgen.org/sc/participating.html) an ongoing international collaborative effort to study the genetic determinants of gene expression changes in immune blood cells from thousands of individuals using a meta-analysis framework. We expanded this statistical framework to study gene expression differences across individuals, including age, sex and genetic ancestry differences across immune cell types.
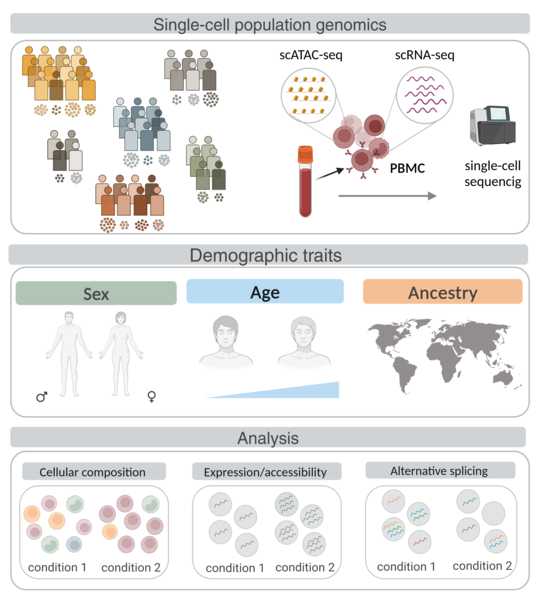
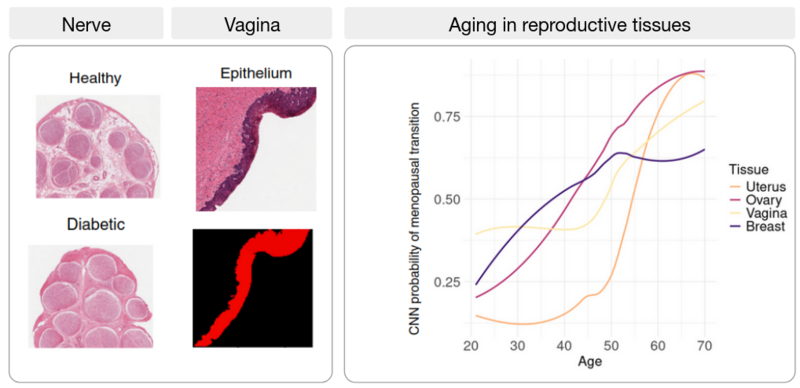
Research line 3: Artificial Intelligence (AI) meets the transcriptome: from tissue architecture to gene expression
The development of artificial intelligence (AI) holds great promise for biomedicine: AI has been on the rise for image-based diagnosis, health risk detection, and personalized medicine. In this line of research, we aim to combine histopathological image analysis using AI with transcriptomic data to find associations between tissue architecture and gene expression changes during disease progression. Applying image classification algorithms to nerve images from healthy and diabetic patients, we find that the nerve fascicles are enlarged in diabetic patients and we uncover novel target genes associated with diabetic neuropathy. Similarly, we identify changes in thyroid follicles associated with cigarette smoking, and, in atherosclerotic patient images, we trace artery wall thickening and calcification progression to their associated genes. By combining transcriptome data and histological image analysis across tissues, we describe differential aging trajectories between organs with corresponding tissue-specific molecular shifts.
Research line 4: Transcriptional regulation of ribosomal RNA and ribosomal proteins
Ribosomes are fundamental units of life and are the main cellular machinery to carry out protein synthesis. Ribosomes are formed by ribosomal proteins and ribosomal RNA, which both have refined regulation at the expression and post-transcriptional level, in an extremely complex assembly process. In recent years, the view of the ribosome has radically changed. Traditionally, the ribosome was viewed as a static cell machinery that carried out protein translation lacking any regulatory roles. Recently, it has been shown that ribosomes can be different between tissues and developmental time points and that these differences may play regulatory roles. Specifically, a handful of studies have suggested that ribosomes can differ in their ribosomal protein composition and this can have an impact on the pool of translated mRNAs. However, the extent of this ribosome heterogeneity and its functional impact is still unclear. In our lab, we recently found evidence that alternative splicing on ribosomal proteins may play a fundamental role in generating ribosome heterogeneity not only between tissues but also between human populations (Garcia-Perez et al. 2023). In addition, we also observed that studies on ribosomal RNA gene expression are extremely scarce, especially in humans. This is mostly due to the fact that ribosomal RNA represents 80% of the RNA in the cell and all RNA sequencing protocols deplete it and treat it as a contaminant. We are developing innovative computational approaches to rescue these discarded ribosomal RNA reads and study gene expression variation in ribosomal RNAs across individuals and tissues.
Group News
Marta Melé is recognized by El Periodico's 25 women to change the world
Marta Melé is awarded the Ancestry Networks for the Human Cell Atlas grant on the project "African ancestry immune cell atlas: genetics, epigenetics and environment". Co-applicant with Sarah Tiskoff (coordinator) and 7 other Principal Investigators. (November 2021)
Raquel Garcia-Perez is awarded the Juan de la Cierva Incorporacion fellowship (August 2021)
Marta Melé is awarded the Plan Nacional Project "The anatomy of the human transcriptome in health and disease" (June 2020)
Marta Melé is awarded a Fundació La Marató Project "Epigenetic Characterization of Cholangiocarcinomas" together with Sandra Peiró (VHIO) (December 2019)
Marta Melé is awarded the L'Oréal-Unesco for Women in Science Award
Kaia Mattioli is awarded Severo Ochoa mobility grant to visit our lab at BSC
